deepEA: a containerized web server for interactive analysis of epitranscriptome sequencing data
Abstract
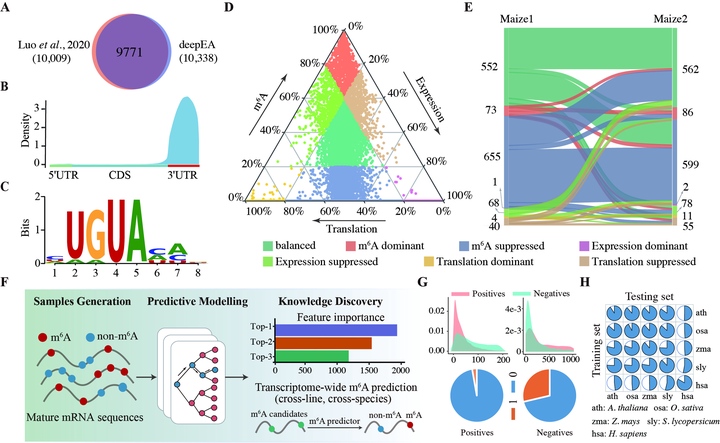
RNA molecules are decorated with a variety of chemical modifications, which make up an epitranscriptome that extensively regulates gene expression and biological processes. With the recent development of high-throughput experimental technologies, epitranscrip- tome sequencing has been a powerful tool to profile dif- ferent types of chemical modifications, such as N6- methyladenosine (m6A), m1A, 5-methylcytidine (m5C), and so on. The resulting epitranscriptome maps reflect dynamic patterns of chemical modifications at a transcriptome-wide scale, resulting in the discovery of reg- ulatory roles in multiple biological processes such as em- bryo development, floral transition, leaf initiation, and stress responses. Integrative pipelines for epitranscriptome sequencing data analysis exist, but these packages were designed for specific or limited functionalities. Moreover, when designing tools for high-throughput sequencing data, ensuring ease of use and reliability become a crucial desideratum. Accordingly, the absence of all-in-one, easy-to-use, and reliable platforms is greatly obstructing in-depth analyses of epitranscriptome sequencing data for both computational and experimental biologists. To address this limitation, we here present deepEA, an interactive web-based platform capable of supporting in-depth analysis of epitranscriptome sequencing data.