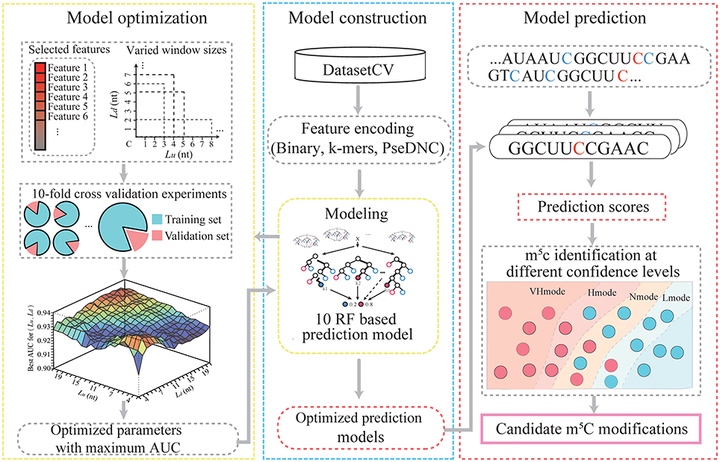
Abstract
The emergence of epitranscriptome opened a new chapter in gene regulation. 5-methylcytosine (m5C), as an important post-transcriptional modification, has been identified to be involved in a variety of biological processes such as subcellular localization and translational fidelity. Though high-throughput experimental technologies have been developed and applied to profile m5C modifications under certain conditions, transcriptome-wide studies of m5C modifications are still hindered by the dynamic and reversible nature of m5C and the lack of computational prediction methods. In this study, we introduced PEA-m5C, a machine learning-based m5C predictor trained with features extracted from the flanking sequence of m5C modifications. PEA-m5C yielded an average AUC (area under the receiver operating characteristic) of 0.939 in 10-fold cross-validation experiments based on known Arabidopsis m5C modifications. A rigorous independent testing showed that PEA-m5C (Accuracy [Acc] = 0.835, Matthews correlation coefficient [MCC] = 0.688) is remarkably superior to the recently developed m5C predictor iRNAm5C-PseDNC (Acc = 0.665, MCC = 0.332). PEA-m5C has been applied to predict candidate m5C modifications in annotated Arabidopsis transcripts. Further analysis of these m5C candidates showed that 4nt downstream of the translational start site is the most frequently methylated position. PEA-m5C is freely available to academic users at: https://github.com/cma2015/PEA-m5C.