Machine learning-based differential network analysis: a study of stress-responsive transcriptomes in Arabidopsis
Abstract
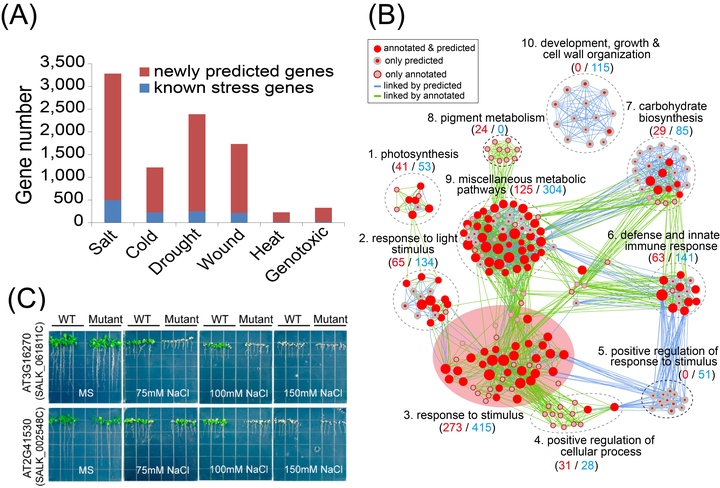
Machine learning (ML) is an intelligent data mining technique that builds a prediction model based on the learning of prior knowledge to recognize patterns in large-scale data sets. We present an ML-based methodology for transcriptome analysis via comparison of gene coexpression networks, implemented as an R package called machine learning-based differential network analysis (mlDNA) and apply this method to reanalyze a set of abiotic stress expression data in Arabidopsis thaliana. The mlDNA first used a ML-based filtering process to remove nonexpressed, constitutively expressed, or non-stress-responsive “noninformative” genes prior to network construction, through learning the patterns of 32 expression characteristics of known stress-related genes. The retained “informative” genes were subsequently analyzed by ML-based network comparison to predict candidate stress-related genes showing expression and network differences between control and stress networks, based on 33 network topological characteristics. Comparative evaluation of the network-centric and gene-centric analytic methods showed that mlDNA substantially outperformed traditional statistical testing-based differential expression analysis at identifying stress-related genes, with markedly improved prediction accuracy. To experimentally validate the mlDNA predictions, we selected 89 candidates out of the 1784 predicted salt stress-related genes with available SALK T-DNA mutagenesis lines for phenotypic screening and identified two previously unreported genes, mutants of which showed salt-sensitive phenotypes.
Chuang Ma
Principal Investigator, Professor, Doctoral Supervisor
My research interests include artificial intelligence, abiotic stress and plant breeding.