Abstract
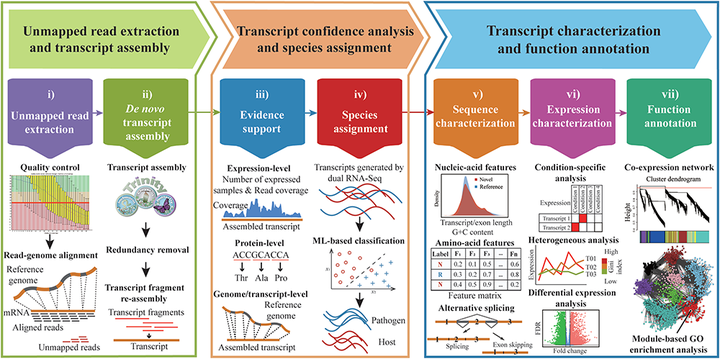
A widely used approach in transcriptome analysis is the alignment of short reads to a reference genome. However, owing to the deficiencies of specially designed analytical systems, short reads unmapped to the genome sequence are usually ignored, resulting in the loss of significant biological information and insights. To fill this gap, we present Comprehensive Assembly and Functional annotation of Unmapped RNA-Seq data (CAFU), a Galaxy-based framework that can facilitate the large-scale analysis of unmapped RNA sequencing (RNA-Seq) reads from single- and mixed-species samples. By taking advantage of machine learning techniques, CAFU addresses the issue of accurately identifying the species origin of transcripts assembled using unmapped reads from mixed-species samples. CAFU also represents an innovation in that it provides a comprehensive collection of functions required for transcript confidence evaluation, coding potential calculation, sequence and expression characterization and function annotation. These functions and their dependencies have been integrated into a Galaxy framework that provides access to CAFU via a user-friendly interface, dramatically simplifying complex exploration tasks involving unmapped RNA-Seq reads. CAFU has been validated with RNA-Seq data sets from wheat and Zea mays (maize) samples. CAFU is freely available via GitHub: https://github.com/cma2015/CAFU.